
Abstract
Currently, the third and fourth waves of the coronavirus disease -19 (COVID-19) pandemic are creating havoc in many parts of the world. Although vaccination programs have been launched in most countries, emerging new strains of the virus along with geographical variations are leading to varying success rates of the available vaccines. The presence of comorbidities such as diabetes, cardiovascular diseases and hypertension is responsible for increasing the severity of COVID-19 and, thus, the COVID-19 mortality rate. Angiotensin-converting enzyme 2 (ACE2), which is utilized by SARS-CoV-2 for entry into host cells, is widely expressed in the lungs, kidneys, testes, gut, adipose tissue, and brain. Infection within host cells mediates RAS overactivation, which leads to a decrease in the ACE2/ACE ratio, AT2R/AT1R ratio, and MasR/AT1R ratio. Such imbalances lead to the development of heightened inflammatory responses, such as cytokine storms, leading to post-COVID-19 complications and mortality. As the association of SARS-CoV-2 infection and hypertension remains unclear, this report provides an overview of the effects of SARS-CoV-2 infection on patients with hypertension. We discuss here the interaction of ACE2 with SARS-CoV-2, focusing on neuronal ACE2 (nACE2), and further shed light on the possible involvement of nACE2 in hypertension. SARS-CoV-2 enters the brain through neuronal ACE2 and spreads in various regions of the brain. The effect of viral binding to neuronal ACE2 in areas of the brain that regulate salt/water balance and blood pressure is also discussed in light of the neural regulation of hypertension in COVID-19.
Similar content being viewed by others

Introduction
The COVID-19 causative agent SARS-CoV-2 not only infects the upper respiratory tract but also spreads to the brain in many patients [1]. Recently, several clinical studies have shown that the presence of one or more comorbidities is associated with the severity of COVID-19 symptoms. Hypertension is recognized as one of the most fatal comorbidities, such as diabetes, cardiovascular diseases, and kidney failure [2,3,4].
The binding of SARS-CoV-2 spike proteins to angiotensin-converting enzyme 2 (ACE2) facilitates its transmission into host cells [5]. These ACE2 proteins are the master regulators of the local and central renin-angiotensin system (RAS). ACE2 catalyzes the conversion of angiotensin I to angiotensin-(1–9) and angiotensin II (Ang II) to angiotensin-(1–7) [6,7,8,9]. An imbalance in the ACE2/ACE ratio can occur due to reduced activity of ACE2 or increased activity of ACE. Consequently, increased AngII induces the production of reactive oxygen species (ROS) and increases the expression of cyclooxygenase enzymes. The increased activity of the ACE/AngII/AT1R axis or the decreased activity of the ACE2/Ang-(1–7)/MasR axis generates pulmonary and neurogenic hypertension and therefore may lead to thrombosis, causing cardiovascular complications culminating in lung damage, heart failure, multiorgan failure and hemorrhage [10, 11]. Recently, clinical studies reported the dissemination of SARS-CoV-2 to other organ systems, including the brain. Clinicians raised concerns that neuropathology inflicted by the virus may also contribute to peripheral effects.
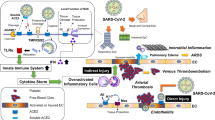
The mechanisms of neuroinvasion by SARS-CoV-2 were obscure until Moriguchi et al. reported the presence of SARS-CoV-2 in the cerebrospinal fluid (CSF) of COVID-19 patients [12]. Neuronal ACE2 (nACE2) is present in different parts of the brain, such as the subfornical organ (SFO), area postrema (AP), paraventricular nucleus (PVN), the dorsal motor nucleus of the vagus (DMNX), nucleus of tractus solitarius (NTS) and rostroventrolateral medulla (RVLM) (Fig. 1A) [13]. The possibility is raised that the presence of nACE2 may play a facilitatory role in the neurotropism of SARS-CoV-2 [14, 15]. However, whether the neuroinvasive and neurotropic capabilities of SARS-CoV-2 owing to the presence of nACE2 in certain parts of the brain contribute to the neural control of hypertension remains to be determined. The overwhelming morbidity due to COVID-19 in patients suffering from heart diseases and hypertension warrants a systematic discussion on the unusual nature of SARS-CoV-2 infection. The clarity in understanding the pathophysiology of SARS-CoV-2-induced neurogenic hypertension is crucial in the development of therapeutic strategies for COVID-19 patient management.

Schematic representation of the effect of SARS-CoV-2 infection on the generation of neurogenic hypertension. A Neuronal ACE2 expression in different parts of the brain, such as the subfornical organ (SFO), area postrema (AP), paraventricular nucleus (PVN), dorsal motor nucleus of the vagus (DMNX), nucleus of tractus solitarius (NTS), and rostroventrolateral medulla (RVLM). B SARS-CoV-2 neuroinvasion is facilitated by its binding to neuronal ACE2 present on neurons, that leads to inflammatory response induction, such as increased secretion of chemokines and cytokines, as well as increased ROS levels. This not only can lead to alterations in neuronal function but also can alter baroreceptor reflex activity, that in turn leads to the development of hypertension
In this review, we summarize the status of knowledge on SARS-CoV-2 infection and discuss the potential of the clinical interplay between nACE2 and SARS-CoV-2 infection in the generation of neurogenic hypertension.
ACE2
In humans, the 40 kb ACE2 gene is located on chromosome Xp22, which comprises of 20 introns and 18 exons. ACE2 is a type I transmembrane protein composed of 805 amino acids. At the N-terminus, ACE2 contains a peptidase domain (PD), and at the C-terminus, a collectrin-like domain is present. Structurally, ACE2 carries only a single catalytic domain that possesses a zinc-binding metallopeptidase motif (HEXXH). ACE2 catalyzes the conversion of angiotensin I into angiotensin-(1–9) [8]. In addition, ACE2 converts angiotensin II to angiotensin-(1–7) [8]. Functionally, this enzyme may exist in two forms: a membrane-bound form and a soluble form. These membrane-bound ACE2 proteins are ubiquitous and are distributed across organs and tissues such as the lung parenchyma, brain, nasal mucosa, lymphoid tissues, testes, renal system and urinary tract [16]. The soluble form of ACE2 is present in a small amount in the blood. This catalytically inactive soluble form of ACE2 is formed by the cleavage of membrane-bound ACE2 by the enzyme disintegrin and metalloproteinase 17 (ADAM17) [17].
ACE2, a receptor for SARS-CoV-2
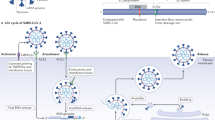
SARS-CoV-2 is a spherical, crown-shaped, enveloped, positive-sense single-stranded RNA virus 66–100 nm in diameter with a genome size of ~29 kb [18]. For host tropism, the S protein of SARS-CoV-2 binds to cell-surface ACE2 proteins through their receptor-binding domain (RBD). Other than ACE2, SARS-CoV-2 has been found to interact with host proteases such as trypsin, factor X, TMPRSS2, furin, cathepsin L, and cathepsin B to facilitate its entry following priming by S protein [5, 19]. It was observed that SARS-CoV-2 RBD has a high binding affinity with the ACE2 than for SARS-CoV [20]. Upon entry into the host, the virus causes the downregulation of membrane-bound ACE2 [21, 22]. This downregulation of ACE2 expression is associated with the elevation of circulating Ang II levels and results in systemic RAS imbalance. Such RAS imbalance is associated with the development of multiple organ damage during SARS-CoV-2 infection [23].
Renin-angiotensin pathway
The renin-angiotensin system (RAS) is an endocrine homeostatic regulator of vascular function that essentially regulates blood volume, blood pressure, urine volume, natriuresis, etc. The RAS is involved in cardiovascular homeostasis owing to its critical role in the regulation of water and electrolyte balance. RAS signaling mechanisms in the vasculature are involved in the maintenance of blood flow. Therefore, the RAS is an integral part of kidney functions. The inactive form of prorenin present in the juxtaglomerular (JG) cells of the kidney undergoes cleavage to produce renin in response to different conditions, such as a decrease in intratubular sodium levels and blood pressure (Fig. 2). The release of renin into the blood causes its reaction with angiotensinogen, a circulating protein. Angiotensinogen, which is produced in the liver and released in the blood, is acted upon by active renin. Free circulating active renin catalyzes the cleavage of angiotensinogen into angiotensin I (Ang I). Furthermore, the angiotensin-converting enzyme (ACE) facilitates the conversion of angiotensin I to angiotensin II (Ang II). Ang II is a biologically active peptide that acts on two types of receptors: Ang II type 1 and type 2 receptors (AT1R and AT2R, respectively). The activation of AT1R by Ang II facilitates vasoconstriction, inflammation, salt and water reabsorption [24]. This effect of Ang II is reduced by angiotensin-converting enzyme 2 (ACE2). ACE2 catalyzes the conversion of Ang II into Ang-(1–7). The resultant heptapeptide binds to G-protein coupled receptors called Mas receptors (MasR). Furthermore, this ACE2/Ang-(1–7)/MasR axis prevents the accumulation of Ang II, a vasoconstrictor, and thus prevents the generation of hypertension [25]. In recent years, the discovery of RAS components in different tissues put forward the concept of local RAS [26]. The RAS components in several parts of the brain serve as a local RAS. All components of the brain RAS, including nACE2, have been identified in neuronal and non neuronal cells, such as the neurons, astrocytes, and microglia of the SFO, PVN, RVLM, AP, and NTS of the brain [27].

Schematic representation of the renin-angiotensin system (RAS). JG cells of the kidney mediate the release of renin. Released renin acts on the liver, produces angiotensinogen, and catalyzes its cleavage into decapeptide angiotensin I (Ang I). Ang I is converted into Ang II through the action of ACE. This Ang II binds to AT1 and AT2 receptors. Ang II binds to AT1 receptors in blood vessels and facilitates vasoconstriction, which increases blood pressure. Ang II also facilitates the release of aldosterone from the adrenal gland, which induces an increase in blood pressure through the retention of water in the kidney. Ang II-AT1R also mediates the secretion of ADH from the pituitary gland, which helps in the absorption of water in collecting ducts and thereby increases blood pressure. ACE2 breaks down Ang II to Ang-(1–7), causing a reduction in Ang II levels and a reduction in blood pressure. ACE Angiotensin-converting enzyme, ACE2 Angiotensin-converting enzyme 2, APA Aminopeptidase A, APN Aminopeptidase N, JG Juxtaglomerular, ADH Antidiuretic hormone, ACE Angiotensin-converting enzyme
Epidemiological findings on the association between hypertension and SARS-CoV-2
During the first wave of the pandemic, it was found that elderly people are at the highest risk of lower respiratory tract infections (LRTIs). However, the new variants of the virus are causing LRTIs in younger populations as well, leading to an increased rate of complications and mortality.
Hypertension is one of the most common comorbidities and is distinctly observed within adult patients. Several clinical studies and epidemiological data from COVID-19 patients link the presence of comorbidities with increased severity of COVID-19, and comorbidities enormously contribute to COVID-19 morbidity. According to the available COVID-19 epidemiological data, the prominent comorbidities that aggravate morbidity include diabetes, cardiovascular diseases (CVDs), cerebrovascular diseases, and hypertension [2,3,4, 23, 28,29,30,31,32,33,34,35,36,37,38] (Table 1). Of these, hypertension was indicated as the most prevalent comorbidity and was associated with an increased mortality rate among COVID-19 patients (Table 1).
According to the available clinical data from COVID-19 patients, immune dysregulation is observed within patients with COVID-19. It is well known that uncontrolled blood pressure stimulates immune system dysregulation. Moreover, in humans, the deregulation of CD4 + and CD8 + T cells plays a causal role in the development of hypertension [39]. In particular, immunosenescent CD8 + T cells are unable to perform antiviral defense effectively and hence lead to the overproduction of cytokines [40, 41]. This cytokine overproduction results in the development of hypertension-related complications such as organ damage [42, 43]. In COVID-19 patients, the elevation of immunomodulatory proteins such as IL2, IL6, IL10, MCP1, and TNF-α in plasma has been observed [44]. Few of these deregulated immunomodulatory proteins associated with the development of hypertension were identified in some experimental models [44]. In addition, immunomodulatory proteins and high levels of plasma troponin T (TnT) and D-dimers have been identified in most patients with severe COVID-19 [30, 33]. Taken together, epidemiological and clinical data from COVID-19 patients suggest that immune dysregulation results in the development of cytokine-storm-induced hypertension and directly correlates with progression to severe COVID-19 and mortality.
Brain, nACE2, and neurogenic hypertension
As recent as two decades ago, the RAS was believed to function with renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin peptides, and their receptors. However, the discovery of ACE2 in 2000 changed the mechanistic understanding of the RAS [8]. Earlier, it was known that Ang II, a vasoactive peptide, is generated through the action of ACE and mediates blood pressure elevation by exerting its action through AT1 receptors. However, the discovery of ACE2 added a twist in the function of the RAS, as it negatively regulates the RAS through the generation of Ang-(1–7) from the degradation of Ang II. ACE2 was identified to balance the vaso-deleterious effect of ACE/Ang II/AT1R [6, 8]. The role of the classical RAS system was found to be associated with the regulation of blood pressure, fluid-electrolyte balance, and systemic vascular resistance. In addition to the classical systemic RAS, the local RAS in the brain also plays a pivotal role in the neural regulation of blood pressure and cardiovascular function. Interestingly, the elements present in brain RAS processes are the same as those present in systemic RAS processes. It has also been noted that brain RAS overactivity, oxidative stress, and cyclooxygenase (COX) are involved in the generation of neurogenic hypertension (Fig. 1B).
ACE2 expression is also ubiquitous in various organ systems, including the brain. Doobay et al. (2007) reported the presence of ACE2 proteins and mRNA in the brains of NSE-AT1A transgenic mice [13]. The isoform of ACE2 in the brain is also identified as neuronal ACE2 (nACE2), that is widely distributed within the brain. Hamming and colleagues observed ACE2 expression within endothelial and smooth muscle cells of the brain [45]. Recently, mouse and human brain transcriptomic analyses revealed a spatial and cell-type distribution of ACE2, calling it nACE2. Excitatory and inhibitory neurons as well as astrocytes and oligodendrocytes also express nACE2 [46]. Of note, mouse dopaminergic neurons and cerebellar cells are nACE2 positives [46]. Notably, the distribution pattern of nACE2 is similar in both mouse and human brains. High nACE2 expression has been found in the substantia nigra and brain vesicles [46].
To understand the role of nACE2 in the brain, different animal model systems have been used (Table 2). Several brain regions were found to be involved in the regulation of BP. Among a few, the prominent area wherein nACE2 was expressed was the hypothalamic paraventricular nucleus (PVN), thereby leading to further speculation on the role of the PVN in the neural control of blood pressure and body fluid homeostasis [13]. In the PVN, nACE2 was particularly detected within presynaptic and vasopressinergic neurons. Ang II infusion in the PVN resulted in the upregulation of AT1R and ACE levels with a subsequent reduction in ACE2 and MAS, leading to hypertension. Proinflammatory cytokines such as TNF-α, IL-6, and IL-1β were also found to be increased in the PVN upon Ang II infusion (Fig. 1B). Sriramula et al. reported that nACE2 overexpression in the PVN of male Sprague–Dawley rats was capable of controlling neurogenic hypertension [47].
Sympathoexcitation has been shown to be associated with the development of hypertension [48]. The RVLM, the vasomotor center, plays an important role in the regulation of the sympathetic nervous system by determining basal central sympathetic outflow. Several studies in stroke-prone spontaneously hypertensive rats (SHRSP) showed that the overexpression of endothelial NOS (eNOS) and neuronal NOS (nNOS) in the RVLM facilitates sympathoinhibition by restoring the impaired baroreflex function [49, 50]. However, it was observed that oxidative stress in the RVLM was involved in the development of neurogenic hypertension in different hypertensive models, such as SHRSP, deoxycorticosterone acetate (DOCA) salt-induced hypertensive models, dietary-induced models, and experimental jet lag models [51,52,53,54]. The rostral ventrolateral medulla (RVLM) is considered to be a relay point because of its role in imparting Ang II signals from the brain to the peripheral nervous system [55]. Yamazato et al. (2007) showed that a spontaneously hypertensive rat (SHR) model exhibited lower ACE2 expression and activity in the RVLM. Lentiviral-mediated ACE2 overexpression within the RVLM was shown to decrease BP in an SHR model [56]. In SHRs, the overexpression of ACE2 in glutamatergic neurons in the RVLM was associated with reduced BP. This suggests that ACE2 per se may not regulate blood pressure. The entire ACE2/Ang-(1–7)/Mas axis contributes to the maintenance of blood pressure [57]. Moreover, nACE2 overexpression in the brain not only increases VEGF, FGF, IFN-γ, and IL6 gene expression but also upregulates eNOS/NO expression, which subsequently decreases the incidence of hypertension-mediated ischemic stroke [58].
The use of deoxycorticosterone acetate (DOCA) salt is one of the most popular ways to induce hypertension in animal models. DOCA induces elevated Ang II levels in the brain without affecting Ang II systemic levels [59]. Increased expression of Nox-2, Nox-4, and nitrotyrosine and reduced expression of SOD and catalases have been observed in a DOCA model of hypertension, causing oxidative stress. Oxidative stress exacerbates reactive oxygen species (ROS) and decreases ACE2 activity through Ang II-mediated stimulation of AT1R. Xia et al. demonstrated that RAS overactivity in DOCA-salt-treated transgenic syn-hACE2 mice increases ADAM17-mediated ACE2 shedding, which creates neurogenic hypertension. ADAM17 knockdown and nACE2 overexpression in transgenic syn-hACE2 mice re-establish baroreflex and autonomic functions and reduce neurogenic hypertension and its associated inflammation [59, 60]. Furthermore, nACE2 overexpression in transgenic syn-hACE2 mice upregulates the AT2 receptor and MasR, thus facilitating a reduction in oxidative stress and COX expression, that attenuates the development of neurogenic hypertension [11]. Conditional overexpression of hACE2 in the brains of syn-hACE2 mice prevented neurogenic hypertension by increasing the AT2R/AT1R and Mas/AT1 receptor ratios [61]. DOCA treatment in synapsin-LoxP-hACE2 (SL) transgenic mice demonstrated brain regions involved in the generation of neurogenic hypertension, and it was observed that the overexpression of hACE2 in the SFO and PVN decreases BP [60].
Effect of SARS-CoV-2 infection in the brain
The first report of the presence of SARS-CoV-2 in CSF grabbed the attention of the scientific community and was a first step toward understanding the neuroinvasive potential and neurotropism mechanism of SARS-CoV-2. Initially, there were several viewpoints on the possible neuroinvasive route of SARS-CoV-2. However, due to the lack of evident research in this area, the answer to this question remains unknown. According to a few reports, SARS-CoV-2 might use a direct or indirect neuroinvasion pathway. The indirect pathway involves the release of the virus from the lungs into the bloodstream, leading to the presentation of viral antigens by antigen-presenting cells to the immune system. These immune cells may cross the blood-brain barrier and facilitate SARS-CoV-2 entry within the CNS through the vagus nerve. A recent study by Rhea EM et al. identified that intranasal administration of the S1 subunit of SARS-CoV-2 into male CD-1 mice facilitated S1 subunit entry within the brain by crossing the BBB [62]. It was also observed that viral infection of endothelial cells in the CNS caused immune system deregulation, which may account for BBB disruption [63]. However, the direct pathway, that is, the transneuronal pathway, involves neuroinvasion through the olfactory nerves of the piriform cortex to the CNS. This pathway permits SARS-CoV-2 entry through primary sensory neuronal cells. After entry, these primary sensory neurons communicate with the neurons of the olfactory system, particularly mitral cells, and thus mediate the transport of the virus from the CSF toward the lymphatic system of the CNS and PNS. An autopsy analysis of COVID-19 patients identified the presence of SARS-CoV-2 in distinct regions of the brain, such as the medulla oblongata, cerebellum, and nasopharynx. In addition, the study suggested that this virus may enter the brain by crossing the neural-mucosal interface [64].
An ultrastructural analysis of autopsy samples from COVID-19 patients confirmed the presence of SARS-CoV-2 in different areas of the CNS, such as the olfactory nerve, gyrus rectus, medulla oblongata, and trigeminal ganglions [65,66,67]. Moreover, a few autopsy studies of COVID-19 patients have shown a loss of neuronal cells and glial cells along with axonal degeneration in the nucleus tractus solitarii, vagus nerve, and dorsal raphe nuclei [65, 67]. Such autopsy analyses in COVID-19 patients confirm the involvement of SARS-CoV-2 in the CNS.
To confirm the pathophysiology of COVID-19, Sun et al. developed a SARS-CoV-2 mouse model in both wild-type C57BL/6 mice and both young and aged hACE2 mice. They injected virus intranasally into transgenic SARS-CoV-2 hACE2 mice and reported the presence of viral S protein expression in neurons, microglial cells, and astrocytes [68]. However, several other studies using K18-hACE2 transgenic SARS-CoV-2 mice identified the presence of the virus in different parts of the brain, such as the thalamus, amygdala, cerebral cortex, hippocampus, medulla, area postrema, hypoglossal nucleus, and midbrain [69,70,71,72]. Altogether, both autopsy studies and experimental analyses support the neuroinvasion and neurotropism of SARS-CoV-2.
Despite neuroinvasion and neurotropism mechanisms, several clinical studies and case report data from COVID-19 patients have revealed the occurrence of neurological manifestations. One such clinical study categorized neurological symptoms into three different categories to understand the neuroinvasive nature of SARS-CoV-2 in patients suffering from [1] CNS symptoms such as headache, dizziness, impaired consciousness, ataxia, epilepsy, and acute cerebrovascular disease; [2] PNS symptoms such as hypogeusia, hyposmia, hypoplasia, and neuralgia; and [3] skeletal muscular symptoms. They identified that ~36.4% of patients were found to manifest neurological symptoms. Among these patients, 25% had CNS symptoms, while the percentages of patients with PNS and skeletal muscle symptoms were 8.9% and 10.7%, respectively. Most of the patients with CNS symptoms had dizziness and headache, whereas hypogeusia was the most common PNS symptom [73]. In a cross-sectional study, the headache was observed to be the most common CNS symptom in COVID-19 patients. Furthermore, in certain patients, the headache was found to be associated with intracranial hypertension [74]. The first case of benign intracranial hypertension (BIH) in a patient with COVID-19 was reported in Brazil [75]. Posterior reversible encephalopathy syndrome (PRES) is triggered by the overactivation of the renin-angiotensin system, which induces hypertension, causes renal injuries, etc. In a case report by Kishfy L et al. in 2020, it was observed that COVID-19 patients with blood pressure fluctuations develop manifestations of hypertensive encephalopathy such as PRES [76].
Another clinical survey found that olfactory taste disorder (OTD) is a clinical symptom of SARS-CoV-2 infection. It is noteworthy that female patients are more susceptible to OTDs than male patients, providing a clue toward sexual differences in the etiology of COVID-19 [77]. Multiple organ failure is another prominent clinical reason for the severity of COVID-19 [78, 79]. In one such report, it was noted that recovered COVID-19 patients were more susceptible to depression, obsessive-compulsive disorder, Alzheimer’s disease, Parkinson’s disease, etc. [80]. In a recent case report, a patient with a SARS-CoV-2 infection also developed a rare type of acute transverse myelitis (ATM) [81].
Most importantly, it is also evident that patients with severe CNS symptoms have low lymphocyte counts, which suggests the presence of immunosuppression. Additionally, the presence of high D-dimer levels is indicative of the development of cerebrovascular disease [73]. It is well known that a lower CD4:CD8 ratio is an indication of a progressive decrease in immunity [82]. However, in a study of COVID-19 patients, the CD4:CD8 ratio was unaltered, but CD8 overexpression was observed, and the number of CD8 + cytotoxic T cells (CTLs) was significantly increased [83]. Cytokine storms are also observed in patients with severe SARS-CoV-2 infections. Such cytokine storms are associated with an impairment of the nicotinic cholinergic neuronal system [84]. CT and MRI studies of patients with SARS-CoV-2 infections suggest the occurrence of a rare complication of acute necrotizing encephalopathy (ANE) [85]. Diffusion tensor imaging (DTI) and 3D T1W1 MRI analyses of recovered COVID-19 patients showed an enlargement of the hippocampus, olfactory cortex, Heschl’s gyrus, and rolandic operculum; a reduction in the mean diffusivity (MD), axial diffusivity (AD), and radial diffusivity (RD); and an increase in fractional anisotropy (FA) in white matter [86]. All these findings suggest that SARS-CoV-2 infection leads to a disruption in the microstructural and functional integrity of the brain [86]. In a recent clinical study, it was observed that some COVID-19 patients without a prior history of hypertension develop hypertension after postinfection. Critical analysis of these data identified elevated levels of cardiac troponin I (cTnl), prolactin, and Ang II in COVID-19 patients [87]. In yet another study, hypertension was identified as the second most prevalent consequence of COVID-19 [88]. Both CNS and PNS symptoms continue to persist even after the infection is cured [89].
Overall, from the critical analysis of published data on COVID-19 patients, one may conclude that neuroinvasion by SARS-CoV-2 causes neuronal degeneration and subsequently activates astrocytes and microglial cells, which generate dysfunction within the cardiovascular center in the brain. Such dysfunction induces RAS imbalance, which in turn results in immune dysregulation. These abnormalities within the CNS after SARS-CoV-2 infection are linked to cellular immune deficiency, which leads to the development of hypertension, cerebrovascular diseases, and coagulopathies.
Treatment strategies to counter SARS-CoV-2-mediated hypertension
SARS-CoV-2 infection in host cells results in RAS overactivation due to the loss of ACE2 either by ADAM17-mediated ACE2 shedding or by increased viral load. Increased Ang II levels lead to increased AT1R activation, which indicates decreases in the ACE2/ACE ratio, AT2R/AT1R ratio, and MasR/AT1R ratio. Such imbalances result in heightened inflammatory responses, such as cytokine storms, leading to post-COVID-19 complications and mortality (Fig. 3A and B). A new line of diagnostics needs to be developed to monitor the inflammatory response and coagulopathy, even in recovering patients, to reduce post-COVID-19 deaths due to cardiac arrest, neuronal hemorrhage, and multiorgan failure. Some of the existing strategies to combat comorbid SARS-CoV-2 infection and hypertension are discussed below.

Effect of SARS-CoV-2 infection on the brain RAS pathway. A In healthy individuals, the brain renin-angiotensin system (RAS) system is well balanced, as blood pressure is well maintained. B However, SARS-CoV-2 infection in patients results in the generation of neurogenic hypertension through the downregulation of the neuronal ACE2/Ang-(1-7)/MasR vasoprotective axis. This downregulation of the vasoprotective axis results in decreases in the ACE2/ACE ratio, AT2R/AT1R ratio, and MasR/AT1R ratio. Vasoprotective axis downregulation also increases cytokine production and neuroinflammation and activates microglial cells. The red arrow indicates the downregulation of enzyme or peptide activity. AT1R Ang II type 1 receptor, AT2R Ang II type 2 receptor, Ang I Angiotensin I, Ang II Angiotensin II, ACE2 Angiotensin-converting enzyme 2, ACE Angiotensin-converting enzyme, MasR Mas receptor
A. Recombinant hACE2
SARS-CoV-2 utilizes ACE2 for entry into host cells. Since the use of rhACE2 treatment may sufficiently prevent the priming of endogenous ACE2 by the S protein of SARS-CoV-2, it may exert beneficial effects by restoring the regulation of the RAS. However, the limitation of this treatment lies in the high molecular size (89.6 kDa) of rhACE2, that may exert lower activity against the tissue RAS. Currently, the developed rhACE2 is in phase II clinical trials [90, 91].
B. AT1 receptor blockers (ARBs) and angiotensin-converting enzyme inhibitors (ACE-is)
ACE-is and ARBs are routinely used for the treatment of hypertension, CVD, and heart failure. The use of an ACE-i exerts a protective effect against high blood pressure by inhibiting the production of Ang II. Several ACE-is, such as lisinopril, captopril, benazepril, fosinopril, and enalapril, are used to treat hypertension. The routine use of an ACE-i causes several adverse side effects, such as dry cough, dizziness, and hyperkalemia. Therefore, ARBs are preferred over ACE-is because they provide excellent efficacy, fewer adverse events, and no negative metabolic effects in patients with hypertension, CVD, and heart failure [90]. From several animal studies, it was evident that peripherally administered ARBs were able to cross the blood-brain barrier (BBB) and block AT1 receptors within the brain, thereby exerting positive effects by lowering blood pressure [54, 92,93,94,95,96]. Although all ARBs are lipophilic to some extent, the variation in their degree of lipophilicity and pharmacokinetic properties, such as plasma half-life, oral bioavailability, and the volume of distribution (Vd), leads to different effects on hypertension. In addition, experimental evidence has shown that ARB liposolubility leads to better absorption and tissue penetration and facilitates sympathoinhibition by strong binding to AT1 receptors [48, 97].
During the early onset of COVID-19, it was hypothesized that ACE inhibitor (ACE-i)/AT1R blocker (ARB) administration may result in the upregulation of ACE2 levels, which decreases Ang II levels and culminates in AT1R response deactivation. This phenomenon might be associated with an increased risk of SARS-CoV-2 infection and disease-linked complications. However, several clinical studies, meta-analyses, single-site cohort studies, and systematic review studies of COVID-19 patients from different countries later reported that the use of ARBs and ACE-is does not have any correlation with an increased risk of SARS-CoV-2 infection. In contrast, the use of ARBs and ACE-is provides beneficial effects, such as a reduction in hospitalization stay and an improved survival rate [98,99,100,101,102,103,104,105,106]. Several societies are working toward the cause of hypertension, such as the International Society of Hypertension, European Society of Hypertension, European Society of Cardiology, Japanese Society of Hypertension, Japanese Circulation Society, and American Heart Association, which support the continuation of ARB/ACE-i treatment in patients with hypertension and those who develop hypertension during infection [107,108,109,110,111]. Currently, several clinical trials on ARBs and ACE inhibitors suggest ARBs as an alternative treatment option for SARS-CoV-2 infection [112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127]. Several such ARBs are still under investigation (Table 3).
From recently available knowledge, it is evident that the progression of SARS-CoV-2 infection is associated with a decrease in ACE2. The resultant increased level of Ang II destabilizes the renin-angiotensin system, causing a decrease in the ACE2/ACE ratio, AT2R/AT1R ratio, and MasR/AT1R ratio, which aggravates the development of cytokine-storm-induced hypertension. The use of ACE-is/ARBs in COVID-19 patients proved to be beneficial and lifesaving. ARBs block the AT1R-Ang II interaction and thereby increase ACE2. However, ACE-is facilitate elevation in ACE2 by blocking the conversion of angiotensin I to angiotensin II. The increase in ACE2 elevates the ACE2/ACE ratio, AT2R/AT1R ratio, and MasR/AT1R ratio and restores the RAS. Overall, ARB/ACE-i treatment in patients with SARS-CoV-2 infections proved to be lifesaving, as it balanced the RAS and lowered the chances of immune dysregulation, which accounted for lower susceptibility to SARS-CoV-2 infection.
Conclusions
SARS-CoV-2 infiltration in the brain has been shown by many clinical studies and supported by autopsy studies. Changes in brain structure have also been shown through CT and MRI. However, further research is warranted to link the process of virus invasion in the brain with CNS symptoms, such as the development of neurogenic hypertension. Based on collective critical evaluation of various scientific letters, viewpoints, discussions, and experimental data, we speculate that, unlike SARS-CoV, SARS-CoV-2 invades the CNS through the blood-brain barrier and through the olfactory nerves of the piriform cortex. In the CNS, the binding of SARS-CoV-2 to ACE2 increases ADAM17, which mediates the shedding of nACE2, culminating in the loss of nACE2; this further causes persistent RAS activation through an increase in the AngII-AT1 axis and generates neurogenic hypertension. As hypertension is convincingly accepted as the most prevalent comorbidity in deceased COVID-19 patients, the development of alternative strategies to maintain the physiological conditions of the RAS to combat SARS-CoV-2 infection cannot be undermined. These alternative strategies should be aimed at preventing the entry of SARS-CoV-2 into host cells and controlling ACE2 shedding and Ang II levels at an early phase of infection. The potential effects of AT1 receptor blockers or soluble ACE2 treatments may be considered for the management of COVID-19.
References
Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain, Behav, Immun. 2020;87:18–22. https://doi.org/10.1016/j.bbi.2020.03.031.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA - J Am Med Assoc. 2020;323:1061–9. https://doi.org/10.1001/jama.2020.1585.
Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical features of 69 cases with coronavirus Disease 2019 in Wuhan, China. Clin Infect Dis. 2020;71:1–9. https://doi.org/10.1093/cid/ciaa272.
Guan WJ, Liang WH, Zhao Y, Liang HR, Chen ZS, Li YM, et al. Comorbidity and its impact on 1,590 patients with COVID-19 in China: A nationwide analysis. medRxiv. 2020. https://doi.org/10.1101/2020.02.25.20027664.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–80. e8. https://doi.org/10.1016/j.cell.2020.02.052.
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–43. https://doi.org/10.1074/jbc.M002615200.
Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–43. https://doi.org/10.1074/jbc.M200581200.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–9. https://doi.org/10.1161/01.res.87.5.e1.
Turner AJ, Hooper NM. The angiotensin-converting enzyme gene family: Genomics and pharmacology. Trends Pharm Sci. 2002;23:177–83. https://doi.org/10.1016/S0165-6147(00)01994-5.
Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circulation Res. 2016;118:1313–26. https://doi.org/10.1161/CIRCRESAHA.116.307708.
Sriramula S, Xia H, Xu P, Lazartigues E. Brain-targeted angiotensin-converting enzyme 2 overexpression attenuates neurogenic hypertension by inhibiting cyclooxygenase-mediated inflammation. Hypertension. 2015;65:577–86. https://doi.org/10.1161/HYPERTENSIONAHA.114.04691.
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. 2020;94:55–8. https://doi.org/10.1016/j.ijid.2020.03.062.
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol - Regul Integr Comp Physiol. 2007;292:R373–81. https://doi.org/10.1152/ajpregu.00292.2006.
Conde Cardona G, Quintana Pájaro LD, Quintero Marzola ID, Ramos Villegas Y, Moscote Salazar LR. Neurotropism of SARS-CoV 2: Mechanisms and manifestations. J Neurological Sci Elsevier B V. 2020;412:116824. https://doi.org/10.1016/j.jns.2020.116824.
Bittmann S A Neurotropic Potential for Brain Damage in COVID-19. Biomed J Sci Tech Res. 2020 Apr 14; 27. https://doi.org/10.26717/bjstr.2020.27.004437.
Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. 2020;16:e9610. https://doi.org/10.15252/msb.20209610.
Xu J, Sriramula S, Xia H, Moreno-Walton L, Culicchia F, Domenig O, et al. Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circ Res. 2017;121:43–55. https://doi.org/10.1161/CIRCRESAHA.116.310509.
Park WB, Kwon NJ, Choi SJ, Kang CK, Choe PG, Kim JY, et al. Virus isolation from the first patient with SARS-CoV-2 in Korea. J Korean Med Sci. 2020;35:e84. https://doi.org/10.3346/jkms.2020.35.e84.
Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78:779 https://doi.org/10.1016/J.MOLCEL.2020.04.022.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–20. https://doi.org/10.1038/s41586-020-2180-5.
Zhou P, Yang XLou, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3. https://doi.org/10.1038/s41586-020-2012-7.
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. https://doi.org/10.1016/j.cell.2020.02.058.
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–74. https://doi.org/10.1007/s11427-020-1643-8.
Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin-angiotensin system: A target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am J Physiol - Heart Circulatory Physiol Am J Physiol Heart Circ Physiol. 2012;302:H1219–30. https://doi.org/10.1152/ajpheart.00796.2011.
Muñoz-Durango N, Fuentes CA, Castillo AE, González-Gómez LM, Vecchiola A, Fardella CE, et al. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int J Mol Sci MDPI AG. 2016;17:797. https://doi.org/10.3390/ijms17070797.
Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system - an endocrine and paracrine system. Endocrinol Endocrinol. 2003;144:2179–83. https://doi.org/10.1210/en.2003-0150.
Davisson RL. Physiological genomic analysis of the brain renin-angiotensin system. Am J Physiol - Regul Integr ComPhysiol. 2003;285:R498–511. https://doi.org/10.1152/ajpregu.00190.2003.
Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med. 2020;18:206 https://doi.org/10.1001/jamacardio.2020.1017.
Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091. https://doi.org/10.1136/bmj.m1091.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020;395:1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Du Y, Tu L, Zhu P, Mu M, Wang R, Yang P, et al. Clinical features of 85 fatal cases of COVID-19 from Wuhan: A retrospective observational study. Am J Respir Crit Care Med. 2020;201:1372–9. https://doi.org/10.1164/rccm.202003-0543OC.
Liu L, Gao JY, Hu WM, Zhang XX, Guo L, Liu CQ, et al. Clinical characteristics of 51 patients discharged from hospital with COVID-19 in Chongqing,China. medRxiv. medRxiv; 2020. 2020.02.20.20025536. https://doi.org/10.1101/2020.02.20.20025536.
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:811–8. https://doi.org/10.1001/jamacardio.2020.1017.
Feng Y, Ling Y, Bai T, Xie Y, Huang J, Li J, et al. COVID-19 with different severities: a multicenter study of clinical features. Am J Respir Crit Care Med. 2020;201:1380–8. https://doi.org/10.1164/rccm.202002-0445OC.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395:497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
Deng Y, Liu W, Liu K, Fang YY, Shang J, Zhou L, et al. Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 in Wuhan, China: A retrospective study. Chin Med J (Engl). 2020;133:1261–7. https://doi.org/10.1097/CM9.0000000000000824.
Fu L, Fei J, Xiang H-X, Xiang Y, Tan Z-X, Li M-D, et al. Analysis of death risk factors among 200 COVID-19 patients in Wuhan, China: a hospital-based case-cohort study. SSRN Electron J. 2020 Mar 24; https://doi.org/10.2139/ssrn.3551430.
Li J, Wang X, Chen J, Zhang H, Deng A. Association of renin-angiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) infection in Wuhan, China. JAMA Cardiol. 2020;5:825–30. https://doi.org/10.1001/jamacardio.2020.1624.
Itani HA, McMaster WG, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, et al. Activation of Human T Cells in Hypertension: Studies of Humanized Mice and Hypertensive Humans. Hypertension. 2016;68:123–32. https://doi.org/10.1161/HYPERTENSIONAHA.116.07237.
Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T clls and C-X-C cemokine receptor type 3 chemokines are increased in human. Hypertension Hypertension 2013;62:126–33. https://doi.org/10.1161/HYPERTENSIONAHA.113.00689.
Mohamed Khosroshahi L, Rezaei N. Dysregulation of the immune response in coronavirus disease 2019. Cell Biology International. Blackwell Publishing Ltd; 2021; Vol. 45:702–7. https://doi.org/10.1002/cbin.11517.
Datta C, Bhattacharjee A. Cytokine storm and its implication in coronavirus disease 2019 (COVID-19). J Immunological Sci. 2020;4:4–21.
Valderrama EV, Humbert K, Lord A, Frontera J, Yaghi S. Severe acute respiratory syndrome coronavirus 2 infection and ischemic stroke. Stroke Lippincott Williams Wilkins. 2020;51:E124–7. https://doi.org/10.1161/STROKEAHA.120.030153.
Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. 2020;71:762–8. https://doi.org/10.1093/cid/ciaa248.
Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7. https://doi.org/10.1002/path.1570.
Chen R, Wang K, Yu J, Howard D, French L, Chen Z, et al. The Spatial and Cell-Type Distribution of SARS-CoV-2 Receptor ACE2 in the Human and Mouse Brains. Front Neurol. 2021;11:573095. https://doi.org/10.3389/fneur.2020.573095.
Sriramula S, Cardinale JP, Lazartigues E, Francis J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res. 2011;92:401–8. https://doi.org/10.1093/cvr/cvr242.
Kishi T. Regulation of the sympathetic nervous system by nitric oxide and oxidative stress in the rostral ventrolateral medulla: 2012 Academic Conference Award from the Japanese Society of Hypertension. Hypertens Res. 2013;36:845–51. https://doi.org/10.1038/hr.2013.73.
Kishi T, Hirooka Y, Ito K, Sakai K, Shimokawa H, Takeshita A. Cardiovascular effects of overexpression of endothelial nitric oxide synthase in the rostral ventrolateral medulla in stroke-prone spontaneously hypertensive rats. Hypertens (Dallas, Tex 1979). 2002;39:264–8. https://doi.org/10.1161/HY0202.102701.
Kishi T, Hirooka Y, Kimura Y, Sakai K, Ito K, Shimokawa H, et al. Overexpression of eNOS in RVLM improves impaired baroreflex control of heart rate in SHRSRostral ventrolateral medulla. Stroke-prone spontaneously hypertensive rats. Hypertens. 2003;41:255–60. https://doi.org/10.1161/01.HYP.0000050649.30821.CB.
Kishi T, Sunagawa K. Experimental “jet lag” causes sympathoexcitation via oxidative stress through AT1 receptor in the brainstem. Annu Int Conf IEEE Eng Med Biol Soc IEEE Eng Med Biol Soc Annu Int Conf. 2011;2011:1969–72. https://doi.org/10.1109/IEMBS.2011.6090555.
Kishi T, Hirooka Y, Ogawa K, Konno S, Sunagawa K. Calorie restriction inhibits sympathetic nerve activity via anti-oxidant effect in the rostral ventrolateral medulla of obesity-induced hypertensive rats. Clin Exp Hypertens. 2011;33:240–5. https://doi.org/10.3109/10641963.2011.583969.
Koga Y, Hirooka Y, Araki S, Nozoe M, Kishi T, Sunagawa K. High salt intake enhances blood pressure increase during development of hypertension via oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertens Res. 2008;31:2075–83. https://doi.org/10.1291/HYPRES.31.2075.
Konno S, Hirooka Y, Kishi T, Sunagawa K. Sympathoinhibitory effects of telmisartan through the reduction of oxidative stress in the rostral ventrolateral medulla of obesity-induced hypertensive rats. J Hypertens. 2012;30:1992–9. https://doi.org/10.1097/HJH.0B013E328357FA98.
Veerasingham SJ, Raizada MK. Brain renin-angiotensin system dysfunction in hypertension: Recent advances and perspectives. Br J Pharm Br J Pharm. 2003;139:191–202. https://doi.org/10.1038/sj.bjp.0705262.
Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension. 2007;49:926–31. https://doi.org/10.1161/01.HYP.0000259942.38108.20.
Wang YK, Shen D, Hao Q, Yu Q, Wu ZT, Deng Y, et al. Overexpression of angiotensin-converting enzyme 2 attenuates tonically active glutamatergic input to the rostral ventrolateral medulla in hypertensive rats. Am J Physiol - Hear Circ Physiol. 2014;307:H182–H90. https://doi.org/10.1152/ajpheart.00518.2013.
Chen J, Zhao Y, Chen S, Wang J, Xiao X, Ma X, et al. Neuronal over-expression of ACE2 protects brain from ischemia-induced damage. Neuropharmacology. 2014;79:550–8. https://doi.org/10.1016/j.neuropharm.2014.01.004.
Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res. 2013;113:1087–96. https://doi.org/10.1161/CIRCRESAHA.113.301811.
Xia H, De Queiroz TM, Sriramula S, Feng Y, Johnson T, Mungrue IN, et al. Brain ACE2 overexpression reduces DOCA-salt hypertension independently of endoplasmic reticulum stress. Am J Physiol - Regul Integr Comp Physiol. 2015;308:R370–8. https://doi.org/10.1152/ajpregu.00366.2014.
Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RAS, et al. Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res. 2010;106:373–82. https://doi.org/10.1161/CIRCRESAHA.109.208645.
Rhea EM, Logsdon AF, Hansen KM, Williams LM, Reed MJ, Baumann KK, et al. The S1 protein of SARS-CoV-2 crosses the blood–brain barrier in mice. Nat Neurosci. 2020;24:368–78. https://doi.org/10.1038/s41593-020-00771-8.
Erickson MA, Banks WA. Neuroimmune Axes of the Blood–Brain Barriers and Blood–Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharm Rev. 2018;70:278. https://doi.org/10.1124/PR.117.014647.
Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. 2021;24:168–75. https://doi.org/10.1038/s41593-020-00758-5.
von Weyhern CH, Kaufmann I, Neff F, Kremer M. Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. Lancet Publ Group. 2020;395:e109. https://doi.org/10.1016/S0140-6736(20)31282-4.
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, et al. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol John Wiley Sons Inc 2020;92:699–702. https://doi.org/10.1002/jmv.25915.
Bulfamante G, Chiumello D, Canevini MP, Priori A, Mazzanti M, Centanni S, et al. First ultrastructural autoptic findings of sars-cov-2 in olfactory pathways and brainstem. Minerva Anestesiologica Edizioni Minerva Med. 2020;86:678–9. https://doi.org/10.23736/S0375-9393.20.14772-2.
Sun SH, Chen Q, Gu HJ, Yang G, Wang YX, Huang XY, et al. A Mouse Model of SARS-CoV-2 Infection and Pathogenesis. Cell Host Microbe. 2020;28:124–133.e4. https://doi.org/10.1016/j.chom.2020.05.020.
Golden JW, Cline CR, Zeng X, Garrison AR, Carey BD, Mucker EM, et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight. 2020;5:e142032. https://doi.org/10.1172/jci.insight.142032.
Yinda CK, Port JR, Bushmaker T, Offei Owusu I, Purushotham JN, Avanzato VA, et al. K18-hACE2 mice develop respiratory disease resembling severe COVID-19. PLOS Pathog. 2021;17:e1009195 https://doi.org/10.1371/journal.ppat.1009195.
Zheng J, Roy Wong L-Y, Li K, Verma AK, Ortiz M, Wohlford-Lenane C, et al. K18-hACE2 mice for studies of COVID-19 treatments and pathogenesis including anosmia. bioRxiv Prepr Serv Biol. 2020. https://doi.org/10.1101/2020.08.07.242073.
Rathnasinghe R, Strohmeier S, Amanat F, Gillespie VL, Krammer F, García-Sastre A, et al. Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. bioRxiv. bioRxiv; 2020. https://doi.org/10.1101/2020.07.06.190066.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020;77:683–90. https://doi.org/10.1001/jamaneurol.2020.1127.
Silva MTT, Lima MA, Torezani G, Soares CN, Dantas C, Brandão CO, et al. Isolated intracranial hypertension associated with COVID-19. 2020 Nov 4; 40:1452–8. https://doi.org/10.1177/0333102420965963.
Noro F, Cardoso FDEM, Marchiori E. COVID-19 and benign intracranial hypertension: A case report. Rev Soc Bras Med Tro. 2020;53:1. https://doi.org/10.1590/0037-8682-0325-2020.
Kishfy L, Casasola M, Banankhah P, Parvez A, Jan YJ, Shenoy AM, et al. Posterior reversible encephalopathy syndrome (PRES) as a neurological association in severe Covid-19. J Neurol Sci. 2020;414. https://doi.org/10.1016/j.jns.2020.116943.
Giacomelli A, Pezzati L, Conti F, Bernacchia D, Siano M, Oreni L, et al. Self-reported olfactory and taste disorders in patients with severe acute respiratory coronavirus 2 infection: A cross-sectional study. Clinical Infectious Diseases. Oxford University Press; 2020;71:889–90.
Collange O, Tacquard C, Delabranche X, Leonard-Lorant I, Ohana M, Onea M, et al. Coronavirus disease 2019: Associated multiple organ damage. Open Forum Infect Dis. 2020; 7. https://doi.org/10.1093/ofid/ofaa249.
Zaim S, Chong JH, Sankaranarayanan V, Harky A. COVID-19 and Multiorgan Response.Current Problems in Cardiology. Mosby Inc.; 2020;45:100618. https://doi.org/10.1016/j.cpcardiol.2020.100618.
Nath A. Neurologic complications of coronavirus infections. Neurol NLM (Medlin). 2020;94:809–10. https://doi.org/10.1212/WNL.0000000000009455.
Shahali H, Ghasemi A, Farahani RH, Nezami Asl A, Hazrati E. Acute transverse myelitis after SARS-CoV-2 infection: a rare complicated case of rapid onset paraplegia. J Neurovirol. 2021;27:354–8. https://doi.org/10.1007/s13365-021-00957-1.
Thornhill J, Inshaw J, Kaleebu P, Cooper D, Ramjee G, Schechter M, et al. Brief report: enhanced normalization of CD4/CD8 ratio with earlier antiretroviral therapy at primary HIV infection. J Acquir Immune Defic Syndr. 2016;73:69–73. https://doi.org/10.1097/QAI.0000000000001013.
Ganji A, Farahani I, Khansarinejad B, Ghazavi A, Mosayebi G. Increased expression of CD8 marker on T-cells in COVID-19 patients. Blood Cells, Mol Dis. 2020;83:102437 https://doi.org/10.1016/j.bcmd.2020.102437.
Farsalinos K, Niaura R, Le Houezec J, Barbouni A, Tsatsakis A, Kouretas D, et al. Editorial: Nicotine and SARS-CoV-2: COVID-19 may be a disease of the nicotinic cholinergic system. Toxicology Reports. Elsevier Inc.; 2020;Vol. 7:658–63. https://doi.org/10.1016/j.toxrep.2020.04.012.
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: Imaging features. Radiol Radiol Soc North Am Inc. 2020;296:E119–20. https://doi.org/10.1148/radiol.2020201187.
Lu Y, Li X, Geng D, Mei N, Wu PY, Huang CC, et al. Cerebral micro-structural changes in COVID-19 patients – An MRI-based 3-month follow-up study: A brief title: cerebral changes in COVID-19. E Clin Med. 2020;25. https://doi.org/10.1016/j.eclinm.2020.100484.
Chen G, Li X, Gong Z, Xia H, Wang Y, Wang X, et al. Hypertension as a sequela in patients of SARS-CoV-2 infection. PLoS One. 2021;16:e0250815. https://doi.org/10.1097/QAI.0000000000001013.
Al-Aly Z, Xie Y, Bowe B High-dimensional characterization of post-acute sequelae of COVID-19. Nat 2021 5947862. 2021; 594:259–64. https://doi.org/10.1038/s41586-021-03553-9.
Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med 2021. 274.2021;27:601–15. https://doi.org/10.1038/s41591-021-01283-z.
Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21. https://doi.org/10.1186/s13054-017-1823-x.
Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52:783–92. https://doi.org/10.1007/s40262-013-0072-7.
Araki S, Hirooka Y, Kishi T, Yasukawa K, Utsumi H, Sunagawa K. Olmesartan reduces oxidative stress in the brain of stroke-prone spontaneously hypertensive rats assessed by an in vivo ESR method. Hypertens Res 2009 3212. 2009;32:1091–6. https://doi.org/10.1038/hr.2009.160.
Leenen FHH, Yuan B. Prevention of Hypertension by Irbesartan in Dahl S Rats Relates to Central Angiotensin II Type 1 Receptor Blockade. Hypertension. 2001;37:981–4. https://doi.org/10.1161/01.HYP.37.3.981.
Nishimura Y, Ito T, Hoe K, Saavedra JM. Chronic peripheral administration of the angiotensin II AT(1) receptor antagonist candesartan blocks brain AT(1) receptors. Brain Res. 2000;871:29–38. https://doi.org/10.1016/S0006-8993(00)02377-5.
Kishi T, Hirooka Y, Sunagawa K. Sympathoinhibition caused by orally administered telmisartan through inhibition of the AT1 receptor in the rostral ventrolateral medulla of hypertensive rats. Hypertens Res 2012 359. 2012;35:940–6. https://doi.org/10.1038/hr.2012.63.
Sueta D, Koibuchi N, Hasegawa Y, Toyama K, Uekawa K, Katayama T, et al. Telmisartan exerts sustained blood pressure control and reduces blood pressure variability in metabolic syndrome by inhibiting sympathetic activity. Am J Hypertens. 2014;27:1464–71. https://doi.org/10.1093/AJH/HPU076.
Michel MC, Foster C, Brunner HR, Liu L. A systematic comparison of the properties of clinically used angiotensin II Type 1 receptor antagonists. Pharm Rev. 2013;65:809–48. https://doi.org/10.1124/PR.112.007278.
Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, et al. Renin–angiotensin–aldosterone system inhibitors and risk of Covid-19. NEJM. 2020;382:2441–8. https://doi.org/10.1056/NEJMOA2008975.
Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors—lessons from available evidence and insights into COVID-19. Hypertens Res 2020 437. 2020;43:648–54. https://doi.org/10.1038/s41440-020-0455-8.
Shibata S, Arima H, Asayama K, Hoshide S, Ichihara A, Ishimitsu T, et al. Hypertension and related diseases in the era of COVID-19: a report from the Japanese Society of Hypertension Task Force on COVID-19. Hypertens Res 2020 4310. 2020;43:1028–46. https://doi.org/10.1038/s41440-020-0515-0.
Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin ii receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. 2020;126:1671–81. https://doi.org/10.1161/CIRCRESAHA.120.317134.
Abajo FJ, de, Rodríguez-Martín S, Lerma V, Mejía-Abril G, Aguilar M, García-Luque A, et al. Use of renin–angiotensin–aldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: a case-population study. Lancet. 2020;395:1705–14. https://doi.org/10.1016/S0140-6736(20)31030-8.
Guo X, Zhu Y, Hong Y. Decreased mortality of COVID-19 with renin-angiotensin-aldosterone system inhibitors therapy in patients with hypertension. Hypertension 2020;11:E13–4. https://doi.org/10.1161/HYPERTENSIONAHA.120.15572.
Khera R, Clark C, Lu Y, Guo Y, Ren S, Truax B, et al. Association of angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers with the risk of hospitalization and death in hypertensive patients with COVID‐19. J Am Heart Assoc. 2021;10:18086 https://doi.org/10.1161/JAHA.120.018086.
Yang G, Tan Z, Zhou L, Yang M, Peng L, Liu J, et al. Effects of angiotensin II receptor blockers and ACE (Angiotensin-Converting Enzyme) inhibitors on virus infection, inflammatory status, and clinical outcomes in patients with COVID-19 and hypertension. Hypertension. 2020;76:51–8. https://doi.org/10.1161/HYPERTENSIONAHA.120.15143.
Kai H, Kai M, Niiyama H, Okina N, Sasaki M, Maeda T, et al. Overexpression of angiotensin-converting enzyme 2 by renin-angiotensin system inhibitors. Truth or myth? A systematic review of animal studies. Hypertens Res 2021 448. 2021;44:955–68. https://doi.org/10.1038/s41440-021-00641-1.
Statement from the American Heart Association the HFS of A and the AC of C. Patients taking ACE-i and ARBs who contract COVID-19 should continue treatment, unless otherwise advised by their physician. American Heart Assoc. 2020;1–3.
International Society of Hypertension. A statement from the International Society of Hypertension on COVID-19. International Society of Hypertension. 2020.
Rosei AE, Simone de G, Gyh L. Statement of the European Society of Hypertension (ESH) on hypertension, Renin-Angiotensin System (RAS) blockers and COVID-19 April 15 th 2020. Cardiovasc Res 2020 Hypertens J Hypertens. 2018;36:1953–2041. https://doi.org/10.1093/cvr/cvaa100.
Simone G de. Position Statement of the ESC Council on Hypertension on ACE-Inhibitors and Angiotensin Receptor Blockers. Eur Soc Cardiol. 2020;2–3.
Kishi T, Hirano T, Mizuno A, Hashimoto Y, Matsumoto C, Fukuda M, et al. Joint Declaration on COVID-19 by the Japan Stroke and Japanese Circulation Societies. Circ Rep. 2020;2:343–4. https://doi.org/10.1253/CIRCREP.CR-20-0040.
Switch of Renin-Angiotensin System Inhibitors in Patients With Covid-19 - ClinicalTrials.gov. NCT04493359. https://clinicaltrials.gov/ct2/show/NCT04493359.
Renin-Angiotensin System Inhibitors and COVID-19. ClinicalTrials.gov. NCT04331574. https://clinicaltrials.gov/ct2/show/NCT04331574.
ACE Inhibitors or ARBs Discontinuation in Context of COVID-19 Pandemic. ClinicalTrials.gov. NCT04329195. https://clinicaltrials.gov/ct2/show/NCT04329195.
Hypertension in Patients Hospitalized With COVID-19. ClinicalTrials.gov. NCT04318301. https://clinicaltrials.gov/ct2/show/NCT04318301.
Long-term use of drugs that could prevent the risk of serious COVID-19 infections or make it worse. ClinicalTrials.gov. NCT04356417. https://clinicaltrials.gov/ct2/show/NCT04356417.
Elimination or prolongation of ACE inhibitors and ARB in coronavirus disease 2019. ClinicalTrials.gov. NCT04338009. https://clinicaltrials.gov/ct2/show/NCT04338009.
Bauer A, Schreinlechner M, Sappler N, Dolejsi T, Tilg H, Aulinger BA. et al.Discontinuation versus continuation of renin-angiotensin-system inhibitors in COVID-19 (ACEI-COVID): a prospective, parallel group, randomised, controlled, open-label trial.Lancet Respir Med. 2021;9:863–72.
Effects of discontinuing renin-angiotensin system inhibitors in patients with and without COVID-19.ClinicalTrials.gov. NCT04351581. https://clinicaltrials.gov/ct2/show/NCT04351581.
Derington CG, Cohen JB, Mohanty AF, Greene TH, Cook J, Ying J, et al. Angiotensin II receptor blocker or angiotensin-converting enzyme inhibitor use and COVID-19-related outcomes among US Veterans. PLoS One. 2021;16:e0248080 https://doi.org/10.1371/journal.pone.0248080.
ARB, ACEi, DRi in COVID-19. ClinicalTrials.gov. NCT04364984. https://clinicaltrials.gov/ct2/show/NCT04364984.
Hakeam HA, Alsemari M, Duhailib ZA, Ghonem L, Alharbi SA, Almutairy E, et al. Association of angiotensin-converting enzyme inhibitors and angiotensin II blockers with severity of COVID-19: a multicenter, prospective study. J Cardiovasc Pharm Ther. 2021;26:244–52. https://doi.org/10.1177/1074248420976279.
Coronavirus (COVID-19) ACEi/ARB Investigation.ClinicalTrials.gov. NCT04330300. https://clinicaltrials.gov/ct2/show/NCT04330300.
Angiotensin receptor blockers and angiotensin-converting enzyme inhibitors and adverse outcomes in patients with COVID19. ClinicalTrials.gov. NCT04364893. https://clinicaltrials.gov/ct2/show/NCT04364893.
Influenza Vaccination, ACEI and ARB in the Evolution of SARS-Covid19 Infection. ClinicalTrials.gov. NCT04367883. https://clinicaltrials.gov/ct2/show/NCT04367883.
Russell JA, Marshall JC, Slutsky A, Murthy S, Sweet D, Lee T, et al. Study protocol for a multicentre, prospective cohort study of the association of angiotensin II type 1 receptor blockers on outcomes of coronavirus infection. BMJ Open. 2020;10:e040768. https://doi.org/10.1136/bmjopen-2020-040768.
ACE Inhibitors, Angiotensin II Type-I Receptor Blockers and Severity of COVID-19. ClinicalTrials.gov. Italy NCT04318418. https://clinicaltrials.gov/ct2/show/NCT04318418.
Acknowledgements
We thank the Indian Council of Medical Research (ICMR), Government of India (GOI), for the award of the Senior Research Fellowship to Mr. Prakash G. Kulkarni (ICMR SRF File no. 2019-6565/CMB/BMS). AJS acknowledges the funding support by the University Grants Commission (UGC), GOI (F.4-5/151-FRP/2014/BSR); the Science and Engineering Research Board (SERB), GOI (EMR/2017/000621); and Rashtriya Uchchatar Shiksha Abhiyan (RUSA), GOI. TB thanks D. Y. Patil Vidyapeeth, Pune for an intramural research grant (DPU/559(1)/2020).
Author information
Authors and Affiliations
Contributions
PGK performed the literature review and drafted the paper. AJS drafted and reviewed the paper. TB drafted and reviewed the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kulkarni, P.G., Sakharkar, A. & Banerjee, T. Understanding the role of nACE2 in neurogenic hypertension among COVID-19 patients. Hypertens Res 45, 254–269 (2022). https://doi.org/10.1038/s41440-021-00800-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-021-00800-4
